 The only treatment for heart valve disease is prosthetic valve replacement. There remain no clinically useful biologically based diagnostics specific to valve disease or biologically based therapies. This is largely due to the limited understanding of how the resident endothelial and interstitial cells of the valve work together to appropriately maintain function. The vast majority of valve biology research focuses on calcification by the valve interstitial cell (VIC). We however have also focused on the valve endothelial cells (VEC), which mediate all biochemical and hemodynamic signals from the blood, and are therefore critical to understand for effecting therapy. VEC have vastly different gene expressions and cellular behaviors than arterial endothelium. Inflammatory activation of VEC is present at the earliest stages of valve disease, but the pathological consequences of this are not known. In advanced stages, VEC monolayers are disrupted at sites of calcific lesions. Untangling these interactions is challenging with purely genetic approaches that necessarily affect both VEC and VIC phenotype the same. Furthermore, these heterogeneous cellular interactions take place within a mechanically active 3D microenvironment that cannot be ignored.
The only treatment for heart valve disease is prosthetic valve replacement. There remain no clinically useful biologically based diagnostics specific to valve disease or biologically based therapies. This is largely due to the limited understanding of how the resident endothelial and interstitial cells of the valve work together to appropriately maintain function. The vast majority of valve biology research focuses on calcification by the valve interstitial cell (VIC). We however have also focused on the valve endothelial cells (VEC), which mediate all biochemical and hemodynamic signals from the blood, and are therefore critical to understand for effecting therapy. VEC have vastly different gene expressions and cellular behaviors than arterial endothelium. Inflammatory activation of VEC is present at the earliest stages of valve disease, but the pathological consequences of this are not known. In advanced stages, VEC monolayers are disrupted at sites of calcific lesions. Untangling these interactions is challenging with purely genetic approaches that necessarily affect both VEC and VIC phenotype the same. Furthermore, these heterogeneous cellular interactions take place within a mechanically active 3D microenvironment that cannot be ignored.Our lab has been tackling these challenges for nearly 20 years. We have made pioneering discoveries into valvular endothelial mechanobiology and their regulation of underlying interstitial cell phenotype. We develop and apply innovative in vitro culture systems to dissect shared and unique mechanisms of endothelial and interstitial cell behavior. These are integrated into novel bioreactors that apply physiological and pathological mechanical forces to elucidate their role in valvular interactions. We further employ genetic manipulation and live cell tracking to elaborate how cell specific molecular regulation affect homeostasis and disease pathogenesis. Together, these approaches help us better understand how these cells respond to changes in their environment, hopefully translating to biologically based strategies to diagnose and prevent valve deterioration, delaying or eliminating the need for prosthetic replacement.
Selected Projects:
Developmental Reactivation in Heart Valve Pathogenesis
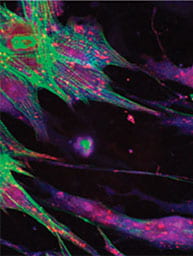
Many of the biological programs that coordinate homeostasis of the valves have their origins in embryonic development. As the tissue matures, these programs abate to a quiescent mode that is poorly understood. During disease pathogenesis, these same molecules and cellular decisions are markedly reactivated, but this time instead of restoring tissue health promote pathogenic remodeling towards fibrosis and calcification. A cardinal example we have focused on is the reactivation of endothelial to mesenchymal transformation in diseased heart valves. We have developed unique in vitro test environments to quantify and profile the phenotypes of endothelium that transform and those that remain quiescent. We have discovered that transformed endothelium is a uniquely calcifying progenitor. We are now identifying novel signaling mediators of these processes, including inflammatory and oxidative stress molecules. We also have identified unique roles for mechanobiological signaling through adhesion proteins. We are testing how small molecule manipulation of these developmentally informed biological programs restore homeostatic VEC-VIC interactions, first in vitro and then in vivo. If successful, we will initiate human trials with these druggable targets with our clinical collaborators. As we identify novel mediators of adult valve homeostasis and calcific degeneration, we will then test for previously unknown roles in embryonic valve formation and maturation. For new mechanisms we identify, we will then explore whether targeted molecular compounds manipulating these shared molecular programs can help reduce malformation severity.
Mechanisms of Advanced Stage Valve Calcification
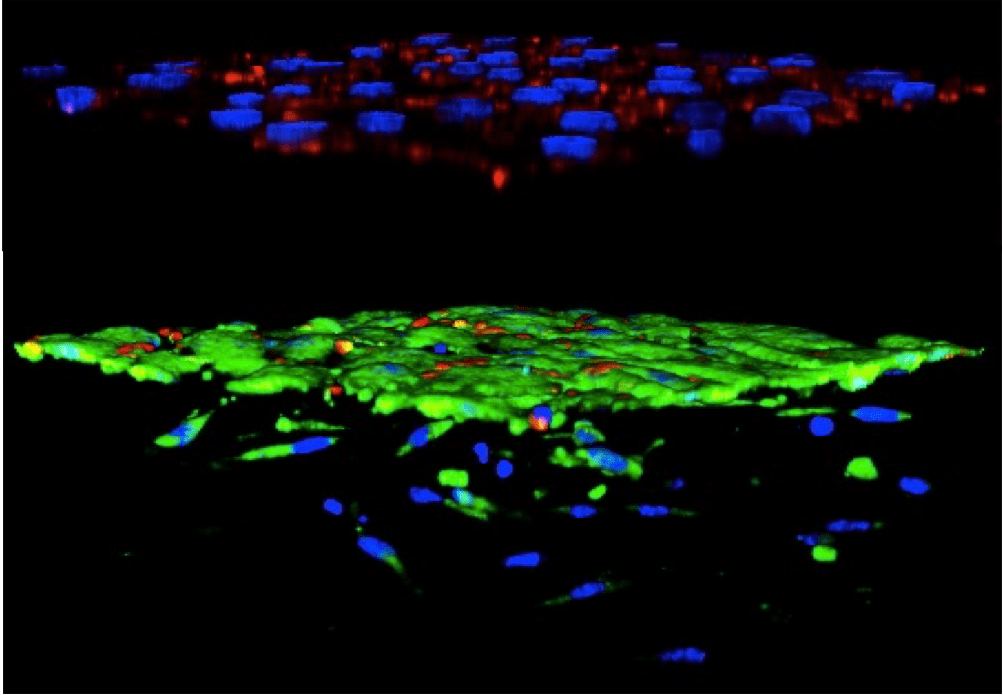
Most research on valve calcification focuses on the early initiating molecular and cellular events. How cells in the valve interact with already existing calcification however is not well understood. Our lab discovered a direct in vivo correlation between long bone loss and accumulation of mineral in the aortic valve via inflammation. We identified that mechanical stress is required for osteogenic differentiation and matrix calcification in 3D cultured VIC, but is inhibited by co-culture with VEC. We discovered that healthy VEC actively maintain the quiescent phenotype of VIC in vivo and in vitro through the secretion of nitric oxide (NO), the first such homeostatic function identified in valves. Diseased VEC (via inflammation and/or oxidative stress) produce less NO, which when combined with inflammatory stimulus results in osteogenic differentiation of VIC and the deposition of calcified matrix. Calcific lesions in the valve contain hydroxyapatite, a mineral found in bone but in valves has distinctly different crystal structure. We have identified a novel correlation between the hydroxyapatite (HA) mineral crystallinity in progressively calcified human valves. We have replicated these different levels of HA crystallinity via engineered HA nanoparticles. We have further innovated a 3D culture approach that creates clinically relevant thickened 3D calcific lesions via cyclic stretch. This provides a unique platform to elucidate molecular and cellular mechanisms in advanced stages of CAVD. We are testing how HA presence and crystallinity independently affect VEC-VIC interactions towards CAVD. We then identify molecular intervention strategies that specifically target these later stage events to delay calcific progression using our established in vitro and in vivo approaches.
Mechanobiology of Heart Valve Matrix Remodeling
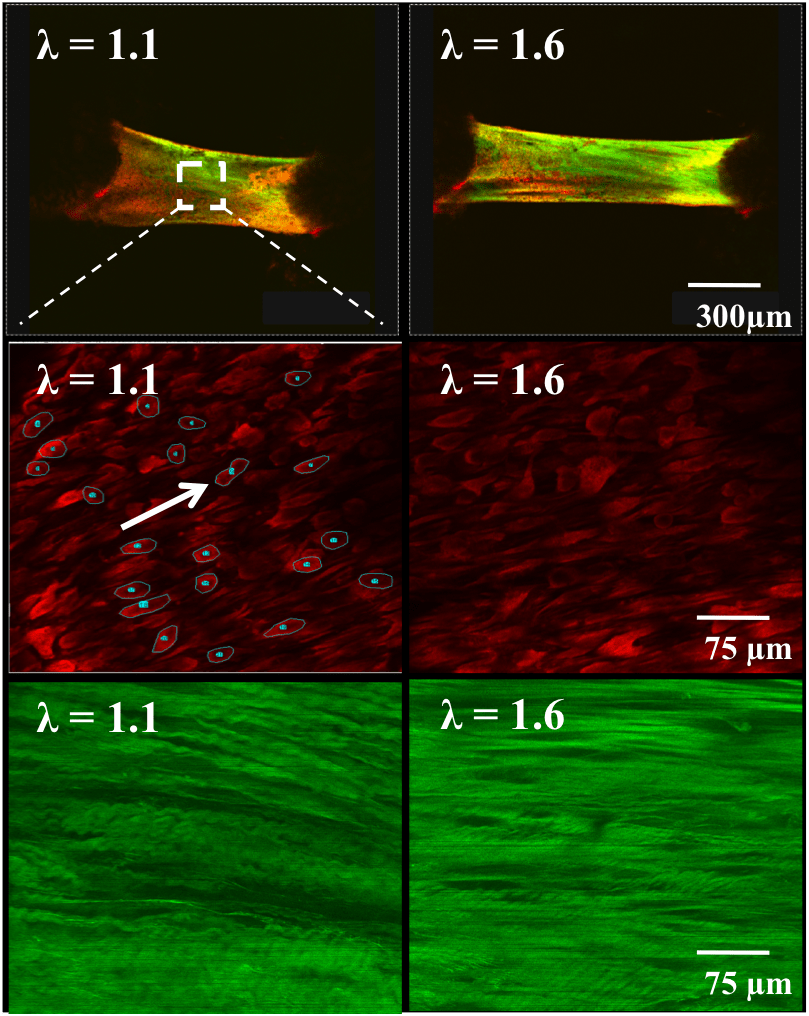
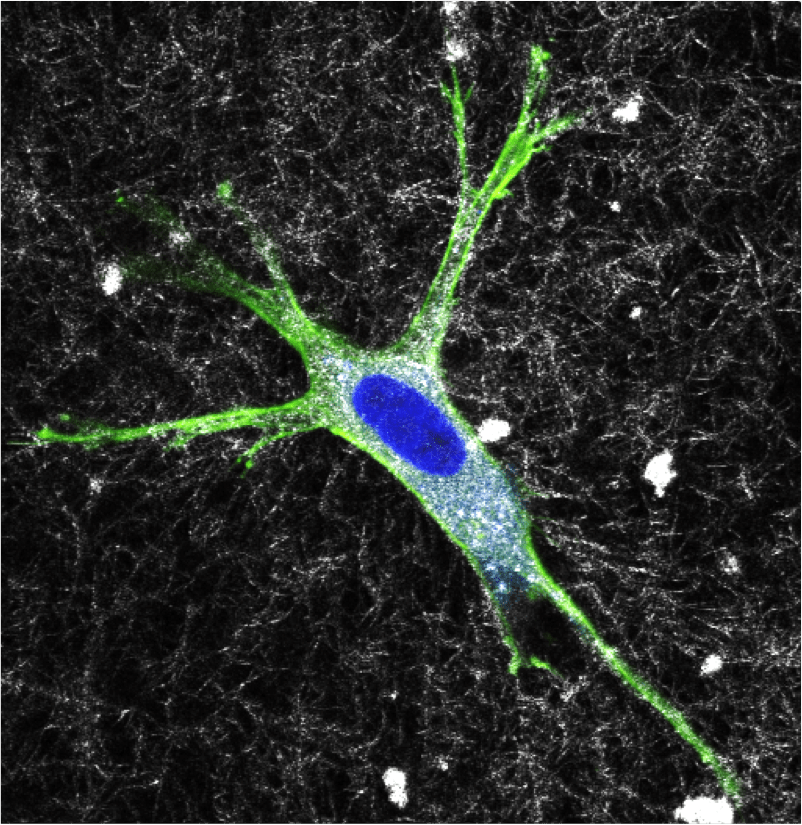
Heart valve leaflets are exposed to a continual barrage of complex mechanical forces that cannot be ignored when studying their biology. Tissue stiffness, cyclic tissue stretch and fluid shear stress affect valve endothelium and interstitial cells in unique and shared ways. We develop and apply unique in vitro bioreactor test beds to evaluate these interactions in 3D in vitro culture and in situ. We developed a unique mechanical analysis system that interrogates subcellular cellular and matrix fiber kinematics in live mouse valve leaflets. We identified that wildtype mouse mitral valves stiffened with age until 4 months, associated with increases in collagen content, fiber alignment, and cellular alignment. Older mitral valves (>10 months) exhibited decreased stiffness associated with disruption in collagen architecture. Interestingly, we determined that Fbn1C1039/+ murine mitral valves (the genetic mutation that causes Marfan syndrome) exhibit reduced stiffness and increased matrix disruption compared to wildtype, indicative of an immature tissue phenotype. We further decipher how the directionality (anisotropy) of mechanical loading affects valvular signaling independently of magnitude or frequency. We discovered that anisotropic strain is a potent driver of cellular reorganization and matrix remodeling. Furthermore, anisotropic strain can uniquely activate signaling activity and affect cell phenotype based on their orientation to the strain field. We also elucidate the mechanotransduction pathways involved, which will open a new path for treating valve disease through restoration of cellular mechanosensitivity.
Noninvasive Diagnosis and Monitoring of Valve Disease Severity
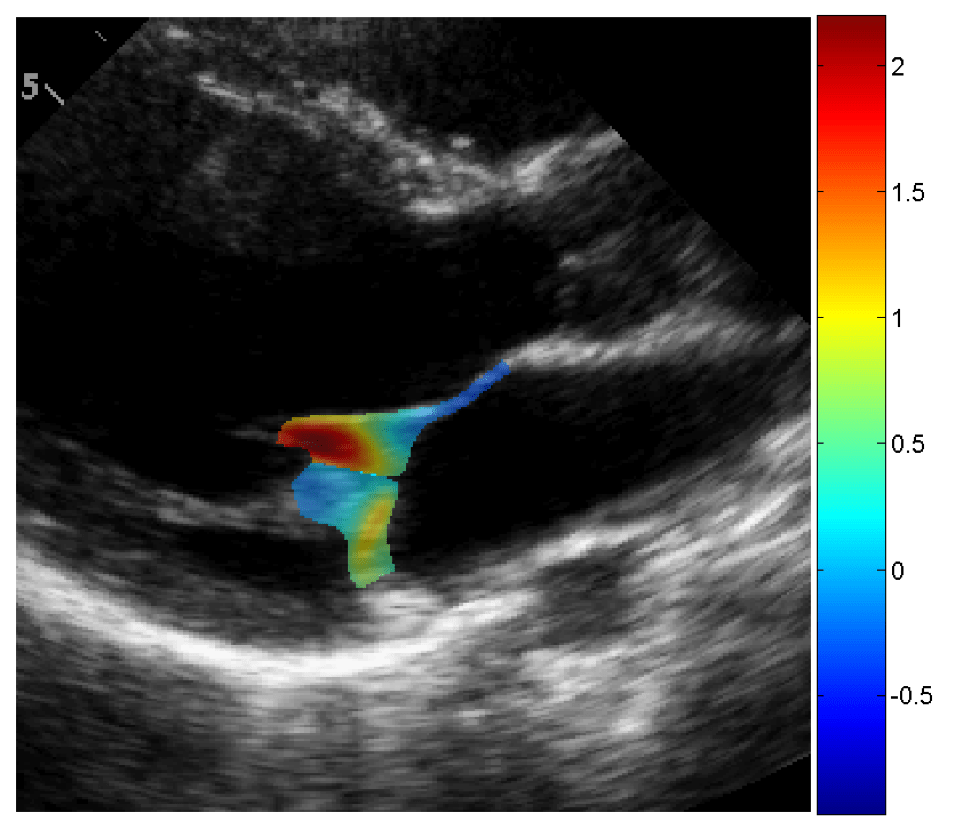
Heart valve disease is diagnosed and monitored using echocardiography and CT imaging. Valve disease progresses very rapidly for some patients, while for others the process can take decades. Cardiologists interpret data across multiple criteria according to their own experience and preferences, limiting standardization and the ability to study disease progression. We have been working with clinical collaborators to create standardized non-invasive indices of aortic valve disease progression, in particular with ultrasound echocardiography. We have retrospectively identified cohorts of patients that exhibit slow or rapid progression of aortic valve disease independently of valve severity, which we are linking to other echocardiographically accessible cardiac parameters. More recently, we have begun prospective analyses that combine imaging with blood testing to develop patient specific profiles of valve function and biological signaling. We are further developing new algorithms to track local valve leaflet deformation kinematics to aid in disease diagnosis and severity monitoring. We have applied these tools to analyze both aortic and mitral valve function, both in humans and in animals that suffer from valve diseases. These tools will aid clinicians in treating patients during the critical window between disease onset and prosthetic replacement.